

Intel® Parallel Computing Center @ RWTH Aachen
Project
 Image: Lammps pictures
Image: Lammps pictures
Molecular dynamics is a highly compute-intensive simulation tool for determining the thermodynamic, kinetic, and transport properties of various materials. The rise of affordable multicore workstations and parallel computing clusters has led to the widespread proliferation of molecular dynamics as a tool through almost all areas of science and engineering, with applications as diverse as protein folding and aggregation to nanoparticle rheology in industrial processing to materials for nuclear waste storage and carbon capture.
In spite of the increasing availability of various accelerator hardware, molecular dynamics codes have been not kept pace, thereby preventing codes from taking full advantage of the latest generation of hardware, including the Intel® Xeon Phi™ line of processors. The goal of our Intel® Parallel Computing Center is to optimize some of the most important computational kernels in the LAMMPS molecular dynamics package for Intel® architectures.
One area which we will address in our work is the problem of multibody potentials, which go beyond the standard pairwise potentials normally used and incorporate three-body, four-body, and sometimes many-body interactions as well. Two such methods that are frequently used in the literature are the AIREBO and Tersoff potentials, which are encountered in simulations of carbon nanotubes, graphene, and other hydrocarbons. By changing how neighbor lists are stored, and by rearranging the data used by the calculations, we will better exploit the intranode parallelism and vectorization capabilities of the Intel® Xeon Phi™ coprocessors.

The most critical part of our work will focus on the long-range solvers present in LAMMPS. In many large-scale molecular simulations, these solvers, which are reponsible for calculating the forces resulting from either electrostatic or dispersion forces, represent the largest computational bottleneck, often accounting for as much as 90 to 95 percent of the total computational expense of a simulation. Consequently, any efficiency gains achieved in these algorithms can have a major impact on the MD community at large. We will exploit the built-in vectorization capabilities of the Intel® Xeon Phi™ by adjusting how data is packed into arrays that handle particle mapping as well as the Poisson solver routines in the particle-particle particle-mesh (PPPM) algorithms in LAMMPS. We will also explore possible improvements in the API which connects LAMMPS to the underlying Fast Fourier transform solvers that drive the PPPM algorithm.
Principal Investigators

Prof. Paolo Bientinesi, Ph.D.
Paolo Bientinesi is Professor for Algorithm-Oriented Code Generation for High-Performance Architectures in the Computer Science department at RWTH Aachen University; he leads the group High-Performance and Automatic Computing within the Aachen Institute for Computational Engineering Science (AICES).
Prof. Bientinesi studied at the University of Pisa (MS, 1998) and at The University of Texas at Austin (Ph.D., 2006); he was a postdoctoral associate at Duke University prior to joining RWTH Aachen. His expertize lies in performance modeling, numerical linear algebra, and automation. He often collaborates with the European Commission as expert evaluator, and is the recipient of the 2009 Karl Arnold Prize from the North Rhine-Westphalian Academy of Sciences and Humanities.

Prof. Ahmed E. Ismail, Ph.D.
Prof. Ahmed E. Ismail leads the Molecular Simulations and Transformations research group, which is affiliated with the Tailor-Made Fuels from Biomass Cluster of Excellence, the Aachener Verfahrenstechnik, and the AICES Graduate School at RWTH Aachen University. Prof. Ismail received his bachelor's and doctorate in chemical engineering from Yale and MIT; after graduation, he was a postdoc and later technical staff member at Sandia National Laboratories.
His research group performs large-scale molecular dynamics simulations for a number of applications, including biomass dissolution, interfacial structure and dynamics, and connections to coarse-grained methods
Articles & Talks
-
LAMMPS' PPPM Long-Range Solver for the Second Generation Xeon Phi
ISC HPC 2017, June 2017
-
Particle-Particle Particle-Mesh (P3M) on Knights Landing Processors
SIAM Conference on Computational Science and Engineering, February 2017
-
The Vectorization of the Tersoff Multi-Body Potential: An Exercise in Performance Portability
SIAM Conference on Computational Science and Engineering, February 2017
-
IPCC @ RWTH Aachen: Optimization of multibody and long-range solvers in LAMMPS
IPCC Showcase, Nov. 2016
-
The Vectorization of the Tersoff Multi-Body Potential: An Exercise in Performance Portability
Slides from the presentation at SC'16, Nov. 2016
-
Accelerating Particle-Particle Particle-Mesh Methods for Molecular Dynamics
Slides from Intel PCC 2016 Fall Forum, Oct. 2016
-
The Vectorization of the Tersoff Multi-Body Potential: An Exercise in Performance Portability
arXiv preprint, SC'16, 2016
-
IPCC @ RWTH Aachen University, Showcase: KNL results
Slides from talk at Intel booth during ISC'16, 2016
-
Molecular Dynamics Research Enhanced by More Scalable LAMMPS HPC Code
Article in Scientific Computing, 2016
-
Optimization of multibody and long-range solvers in LAMMPS
Slides from Intel PCC EMEA Meeting in Ostrava, 2016
-
IPCC @ RWTH Aachen: Optimization of multibody and long-range solvers in LAMMPS
Slides from project showcase, 2016
-
The Tersoff many-body potential: Sustainable performance through vectorization
Article in Proceedings of SC'15 workshop: Producing High Performance and Sustainable Software for Molecular Simulations, 2015
-
The Tersoff many-body potential: Sustainable performance through vectorization
Slides from SC'15 workshop: Producing High Performance and Sustainable Software for Molecular Simulations, 2015
Code on Github
| Repository of code for the Tersoff potential's optimization |
| Repository of code for the Buckingham potential's optimization |
Additional Material
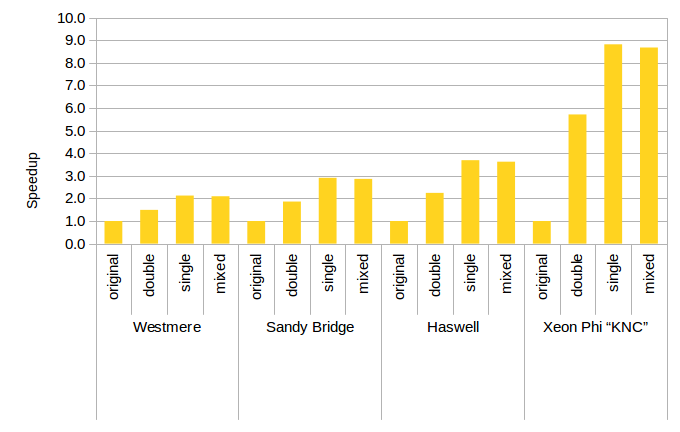 Figure 1: Speedup From Optimization on Various Architectures.
Our optimizations for the Tersoff potential translate to appreciatable gains in performance in real applications.
This benchmark is derived from a simulation of Carbon-Nanotubes.
The optimizations halve the computing time on a wide variety of hardware platforms.
On modern hardware, such as the Xeon Phi coprocessor, these speedups can be dramatical, cutting the runtime by a factor of nine.
Figure 1: Speedup From Optimization on Various Architectures.
Our optimizations for the Tersoff potential translate to appreciatable gains in performance in real applications.
This benchmark is derived from a simulation of Carbon-Nanotubes.
The optimizations halve the computing time on a wide variety of hardware platforms.
On modern hardware, such as the Xeon Phi coprocessor, these speedups can be dramatical, cutting the runtime by a factor of nine.
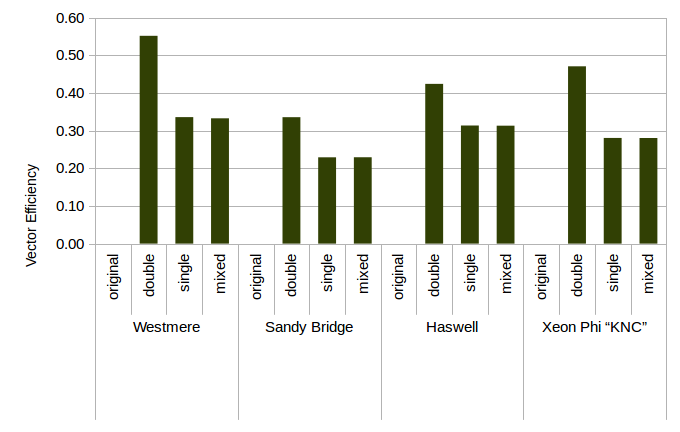 Figure 2: Vector Efficiency From Optimization on Various Architectures.
We use a vectorization framework that makes it possible to estimate the efficiency with which our code uses the vector unit of the hardware.
This measure is relative to the inert parallelism in the vector unit.
One would expect the vector efficiency to decrease, the longer the vectors get.
While this is true within a certain platform, it is notably not true in comparison with each other: The Xeon Phi has 4 times longer vectors than a Westmere-generation processor, but both are even on the efficiency.
This figure is based on the same data as Fig. 1. Note how low effiency does not necessarily translates to low speedups.
Figure 2: Vector Efficiency From Optimization on Various Architectures.
We use a vectorization framework that makes it possible to estimate the efficiency with which our code uses the vector unit of the hardware.
This measure is relative to the inert parallelism in the vector unit.
One would expect the vector efficiency to decrease, the longer the vectors get.
While this is true within a certain platform, it is notably not true in comparison with each other: The Xeon Phi has 4 times longer vectors than a Westmere-generation processor, but both are even on the efficiency.
This figure is based on the same data as Fig. 1. Note how low effiency does not necessarily translates to low speedups.
Team
| Rodrigo Canales | ||
| Markus Höhnerbach | ||
| William McDoniel |
Links
- RWTH Aachen
- AICES - Aachen Institute for Advanced Study in Computational Engineering Science
- Molecular Simulations and Transformations
- LAMMPS
- Intel® Parallel Computing Centers